In a hospital, a patient is fighting a battle against an infectious microbe, and is taking antibiotics to win this battle. Yet as soon as the poisons find their way into the interior of the bug, they are jettisoned out of the cell, making them useless on this occasion. In another setting, a breast cancer patient is fighting a battle against cancer because the tenaciously dividing cancer cells in her body are spitting out anticancer drugs back into her blood stream, preventing them from attacking her tumour. The cancer cells use the same molecular gadgetry as those present in microorganisms. Nature’s common currency is a protein called a 'multidrug' transporter. Embedded within a cell’s plasma membrane, this protein protects a cell by ejecting a variety of molecules - in many cases, toxins - on contact. With cancer cancer cells, the toxins are anticancer agents. The cell might also be a microbe, in which case the toxins are antibiotics.
We live in an era where antibiotic resistance among pathogenic microorganisms has spread at an alarming rate. The World Health Organization states that antibiotic resistance is one the three greatest threats to human health. There are so many interesting questions about multidrug transporters. Given the greatly different three-dimensional structures of multidrug transporters in the various protein families, what allows these systems to catalyze drug efflux reactions with overlapping drug selectivities? How is polyspecificity achieved? Can multidrug transporters be selectively inhibited so that existing antibiotics and anticancer drugs can still be used in the treatment of diseases? Can we develop new antibiotics and anticancer agents that bypass recognition by multidrug transporters? In this lab, we work on answers to these questions. Multidrug transporters are important contributors to clinical drug resistance but are not just for resistance. Selection for drug resistance can also potentially occur in the absence of antibiotics due to the native physiological roles that multidrug transporters can have. We therefore study the interactions of multidrug transporters with 'natural' substrates to define these physiological roles and underlying mechanisms of transport. Our interest in lipid transport is one example of this.
Our methods use several different sources of information including biochemistry, cell biology, and biophysics, and involve techniques such as DNA cloning, gene expression, molecular modelling, site-directed mutagenesis, protein purification and reconstitution, drug binding and transport using radiolabelled and fluorescent substrates, and fluorescence spectroscopy techniques. We study multidrug transporters that are members of different families of membrane proteins, that originate from various pathogenic microorganisms and human tumour cells, and that are structurally different but have a conserved function. It is an exciting research area to work in, and research group to be part of.
The following sections give an overview of some of our projects:
Studies on ABC multidrug transporters
ABC (ATP-binding cassette) transporters comprise one of the largest families of structurally related membrane proteins, and mediate substrate transport in a reaction that is driven by ATP-binding and hydrolysis.
Multidrug/lipid transporter MsbA
The ABC transporter MsbA is expressed in the plasma membrane of many Gram-negative bacteria including pathogens such as Salmonella typhimurium, Vibrio cholerae and Pseudomonas aeruginosa. MsbA is thought to transport lipid A, which is the lipid anchor of lipopolysaccharides in the outer membrane, and which is a potent activator of innate immunity in mammals via Toll-like receptors. In a study on the kinetics of drug and lipid-A binding/transport by MsbA from Escherichia coli, we found that both types of substrates can interact with MsbA with a comparable affinity, and that lipid-A binding can inhibit drug binding. Hence, the lipid environment of a multidrug transporter can be an important factor in its drug selectivity.
Mechanisms of drug and lipid transport. We established in 2003 that E. coli MsbA is a polyspecific transporter capable of recognizing and transporting a wide range of amphiphilic molecules that have the ability to partition in the inner leaflet of the membrane and that are typical substrates of multidrug efflux pumps. This finding was consistent with MsbA’s homology to ABC multidrug transporters such as the human multidrug resistance P-glycoprotein ABCB1 and LmrA in gram-positive Lactococcus lactis. We demonstrated in 2008 that the recognition of small molecule drugs in the MsbA dimer occurs in the vicinity to TMH 6 and 6’ in the central chamber that is formed in the inward-facing conformation near the inner membrane leaflet at the interface between the two half-transporters. In cysteine-cross-linking studies in 2013 we demonstrated that the binding of drugs or Lipid A can initiate dimerization of the nucleotide-binding domains. This dimerization facilitates nucleotide binding, allowing us to carefully measure binding affinities for a variety of nucleotides. We also discovered for the first time in this work that the accessibility of the chamber changes from inward-open to occluded in response to the ATP binding; the latter state can be simultaneously cysteine cross-linked at internal and external loop regions, and exhibits a significantly elevated binding affinity for drugs compared to the inward-facing state. In collaboration with researchers at Imperial College, we further worked on the characterisation of the occluded state in ABC exporters in work focussing on the MsbA homologue McjD, a peptide transporter in E. coli. In a publication in 2016 on MsbA, we presented evidence for the dual mode of energy coupling in the transport of amphiphilic drugs. The transport reaction is driven by conventional ATP binding and hydrolysis and is further facilitated by the chemical proton gradient that exists across the plasma membrane of the cell. We were able to study the two modes of energy coupling in vitro by comparing the transport properties of full-length protein (ATP dependence and ion coupling) with the properties of a drug transport-active truncated transporter that contains the membrane domains but lacks the nucleotide binding domains. These studies on MsbA extend earlier observations on the ABC exporter LmrA in which we demonstrated ion-coupled drug transport by the membrane domains.
Mechanisms of inhibition of MsbA activity. As MsbA is an essential lipid transporter in many gram-negative bacteria (including pathogenic organisms), we also focus on biochemical and structural mechanisms by which MsbA can be inhibited by novel compounds. Recently, we used genetic engineering to tweak a critical part of MsbA – a group of four helices embedded in the transporter (referred to as the tetrahelix bundle) (Figure 1) that acts as a ‘spring-lock’ for the transition from the inward-facing to outward-facing shape. By manipulating the msbA gene, we were able to produce bacteria that made MsbA mutants. We then built the modified proteins into 'inside-out' membrane vesicles in which the transporters import the drug from the outside, thus making it easier to follow the transport process. We were able to show that both transport and the associated changes in protein conformation were greatly impaired by the structural alterations in the tetrahelix bundle, while other features, such as the transporter’s ability to bind ATP and substrate, were not significantly affected. Tetrahelix bundles are shared by all ABC exporters in eubacteria and eukaryotes, and are essential for their activity. As their primary amino acid sequences are different, the tetrahelix bundle might provide an attractive drug target for modulation of ABC transporter activity. See Research at Cambridge for further information.
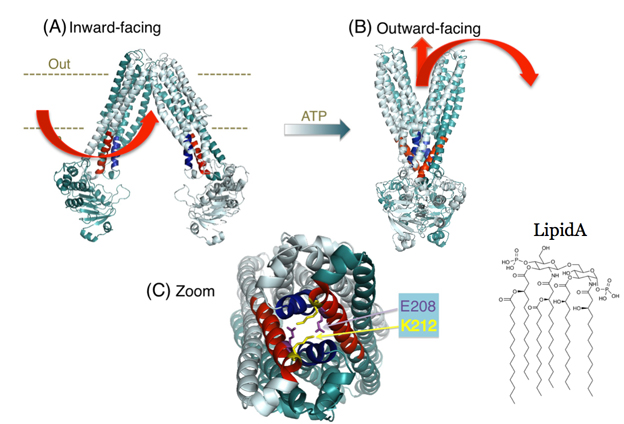
Figure 1. Tetrahelix bundle in the MsbA dimer. (A,B) Proposed conformational changes of the MsbA transporter in the transition from the inward-facing shape (A) to the outward-facing shape (B). Current mechanistic models for substrate transport by the MsbA dimer suggest substrate (Lipid A and drug) binding to the inward-facing conformation followed by a transition to the ATP-bound, outward-facing structure with dimerized nucleotide-binding domains (NBDs), from which Lipid A and drugs are released (arrows in red). (C) Zoomed-in snapshot of the tetrahelix bundle in (B) (in red and blue) viewed from the cytoplasmic side showing examples of stabilizing side-chain interactions.
As the transport activity of MsbA is essential for lipopolysaccharide export and viability of most Gram-negative pathogenic bacteria including Escherichia coli, Salmonella spp., Pseudomonas aeruginosa, Vibrio cholerae, Klebsiella pneumoniae, Yersinia pestis and Acinetobacter baumannii, we also focus on disruption of this activity by novel peptide-based inhibitors that facilitate the development of next-generation antibiotics. This part of our work is done in collaboration with Prof. David Spring at the Department of Chemistry in Cambridge.
Multidrug/lipid transporter LmrA
The ABC multidrug transporter LmrA from Lactococcus lactis is a homologue of the human multidrug resistance P-glycoprotein (ABCB1), another major drug pump involved in drug resistance of human cancer cells. LmrA and ABCB1 have very similar specificities for chemotherapeutic drugs and modulators. LmrA can even functionally substitute for ABCB1 in human lung fibroblast cells. Although ABC transporters are generally thought to be unidirectional extrusion systems, we discovered that LmrA contains a reversible pathway for drugs. Moreover, we demonstrated that if the membrane-embedded, drug-binding part of the transporter is decoupled from the cytosolic part that breaks down ATP to provide metabolic energy (the 'engine' of the transporter) then LmrA acts to import drugs rather than export them. Interestingly, we found that the membrane-embedded part of LmrA can mediate an ion-coupled transport reaction, which points to a functional link between this ABC transporter and secondary-active transporters (see below). Our findings open up the possibility to use LmrA-like transporters for drug delivery in cells. In recent work on LmrA, we dissected the detailed mechanisms underlying ion-coupling in electrophysiological and transport studies using planar lipid bilayers, giant unilamellar vesicles and proteoliposomes, and identified an ion-drug exchange reaction which, upon addition of nucleotides, is switched on in the ATP-bound state and switched off in response to ATP hydrolysis. We propose that this ion exchange reaction is part of the overall transport mechanism for hydrophobic drugs and lipids that are transported by LmrA within the phospholipid bilayer.
Human ABC transporters
In our work on human ABC transporters we study the Breast Cancer Resistance Protein (ABCG2) and the liver bile salt export pump (ABCB11). Our work on ABCG2 revealed the presence of a steroid hormone-binding element in the transporter. As this element modulates ABCG2 activity, its further characterization might provide an opportunity for the development of new therapeutic ligands for ABCG2 in a clinical setting. In our work on the bile salt export pump, we are particularly interested in the link between ABCB11 activity and drug-induced liver injury (DILI). The unpredictability of DILI is one of the most frequent causes of safety-related drug withdrawal with associated delays in our ability to treat human disease. ABCB11 is a homologue of the multidrug resistance P-glycoprotein ABCB1, and is known to interact with many drugs that are structurally and functionally unrelated to bile acids. Various techniques are used in this laboratory to investigate structure-function relationships that underlie the ability of ABCB11 and ABCG2 to interact with a variety of ligands.
Comparison with secondary-active multidrug transporters is interesting
Our research group also studies the molecular properties of secondary-active multidrug transporters, which mediate drug efflux in an antiport reaction that is driven by the transmembrane electrochemical gradient of protons and/or sodium ions. Ion-coupled multidrug transporters can be classified into various families including the Major Facilitator Super (MFS) family, the Resistance-Nodulation-cell Division (RND) family, and the multidrug and toxic compound extrusion (MATE) family. Even though members of these secondary-active transporter families are structurally different from each other and different from ABC exporters, they all show an ability to transport structurally dissimilar drugs. This phenomenon raises interesting questions.
Energetics of multidrug transport by the MFS member LmrP
LmrP is a MFS multidrug transporter from L. lactis that represents a common antibiotic efflux pump in bacteria. Previous work has indicated that ethidium, a monovalent cationic substrate, is exported by LmrP by electrogenic antiport with two (or more) protons. This observation raised the question whether these protons are translocated sequentially along the same pathway, or through different routes. To address this question we constructed a 3-D homology model of LmrP based on the high-resolution structure of the glycerol-3P/Pi antiporter GlpT from E. coli. Interestingly, LmrP is predicted to contain an internal cavity formed at the interface between the two halves of the transporter (each containing 6 transmembrane helices). On the surface of this cavity lie two clusters of polar, aromatic and carboxylate residues with potentially important function in proton shuttling and interactions with cationic drugs (Figure 2). We tested by mutagenesis the possible proton conduction points suggested by this model, and found that there are two proton conduction pathways at play in the drug transport mechanism, of which one might be dedicated to proton transport and the other to drug transport. Proton translocation pathways in LmrP are subject of further research.
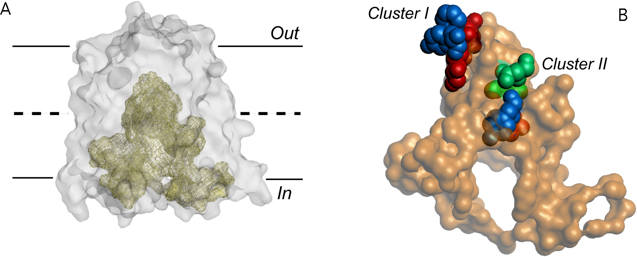
Figure 2. Internal cavity in the proton motive force-dependent MFS member LmrP. (A) The predicted LmrP structure represents an inward facing conformation, exposing an internal cavity to the inner membrane leaflet. Out and In refer to the outside and inside of the plasma membrane, respectively. (B) Residues lining the surface of the internal cavity. Carboxylates (in red) exposed at this surface are organized into two distinct clusters containing additional neutral-polar (in green) and aromatic residues (in blue). The role of these clusters in proton and drug transport by LmrP are being investigated.
Mechanisms of RND multidrug transporters
RND multidrug transporters are important for antibiotic resistance in Gram-negative pathogenic bacteria, where they interact with a membrane fusion protein and outer membrane porin to enable transport from the periplasm across the outer membrane. Even though X-ray crystallography has provided many insights in the structures of the components of RND transporters, much less information is available about the question how these components work together at the functional level.
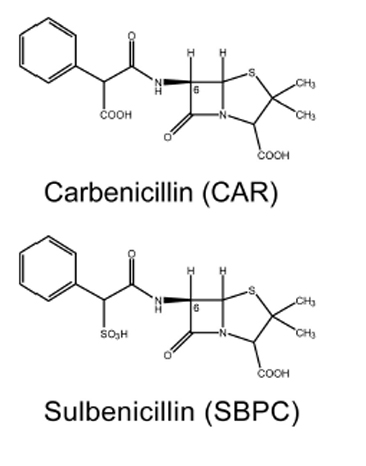 In a collaborative work with Prof. Satoshi Murakami at the Tokyo Institute of Technology, we investigated how β-lactams are recognized by the AcrA-AcrB-TolC system in E. coli. β-Lactam antibiotics are mainstream antibiotics that are indicated for the prophylaxis and treatment of bacterial infections. The AcrA-AcrD-TolC multidrug efflux system confers much stronger resistance on E. coli to clinically relevant anionic β-lactam antibiotics (such as carbenicillin and sulbenicillin) than the homologous AcrA-AcrB-TolC system. Using an extensive combination of chimeric analysis and site-directed mutagenesis, we searched for residues that determine the difference in β-lactam specificity between AcrB and AcrD. We identified three crucial residues at the “proximal” (or access) substrate binding pocket. The simultaneous replacement of these residues in AcrB by those in AcrD (Q569R, I626R, and E673G) transferred the β-lactam specificity of AcrD to AcrB (Figure 3). Our findings indicate for the first time that the difference in β-lactam specificity between AcrB and AcrD relates to interactions of the antibiotic with residues in the proximal binding pocket.
In a collaborative work with Prof. Satoshi Murakami at the Tokyo Institute of Technology, we investigated how β-lactams are recognized by the AcrA-AcrB-TolC system in E. coli. β-Lactam antibiotics are mainstream antibiotics that are indicated for the prophylaxis and treatment of bacterial infections. The AcrA-AcrD-TolC multidrug efflux system confers much stronger resistance on E. coli to clinically relevant anionic β-lactam antibiotics (such as carbenicillin and sulbenicillin) than the homologous AcrA-AcrB-TolC system. Using an extensive combination of chimeric analysis and site-directed mutagenesis, we searched for residues that determine the difference in β-lactam specificity between AcrB and AcrD. We identified three crucial residues at the “proximal” (or access) substrate binding pocket. The simultaneous replacement of these residues in AcrB by those in AcrD (Q569R, I626R, and E673G) transferred the β-lactam specificity of AcrD to AcrB (Figure 3). Our findings indicate for the first time that the difference in β-lactam specificity between AcrB and AcrD relates to interactions of the antibiotic with residues in the proximal binding pocket.
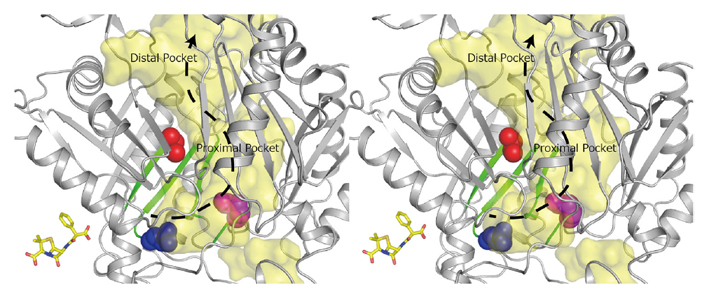
Figure 3. RND efflux pumps are crucial for the intrinsic resistance of Gram-negative bacteria to antibiotics, detergents and toxic ions. Stereo view of the regions in AcrB important for recognition of negatively charged β-lactam antibiotics. Substrate translocation pathway (in yellow) including proximal and distal substrate binding pockets, and direction of substrate transport (dashed arrow) are indicated. Green-colored β-sheet sequences indicate regions that determine the difference in substrate specificity between AcrB and AcrD. The important residues (Gln569, Ile626, and Glu673) for recognition of negatively charged β-lactams are shown in CPK representation. The space colored in yellow is the solvent accessible inner cavity of the AcrB molecule. To put the size of the substrate translocation pathway in AcrB in perspective, a stick model of the β-lactam antibiotic carbenicillin is included at the same scale.
Energy coupling in MATE multidrug transporters
Membrane transporters belonging to MATE family mediate the efflux of unrelated pharmaceuticals from the interior of the cell in organisms ranging from bacteria to human. These proteins are thought to fall into two classes that couple substrate efflux to the influx of either Na+ or H+. Recently, we studied the energetics of drug extrusion by NorM from Vibrio cholerae (Figure 4) in proteoliposomes in which purified NorM protein was functionally reconstituted in an inside-out orientation. We established that NorM simultaneously couples to the sodium-motive force and proton-motive force, and biochemically identified protein regions and residues that play important roles in Na+ or H+ binding. As the positions of protons are not available in current medium and high-resolution crystal structures of MATE transporters, our findings add a previously unrecognized parameter to mechanistic models based on these structures.
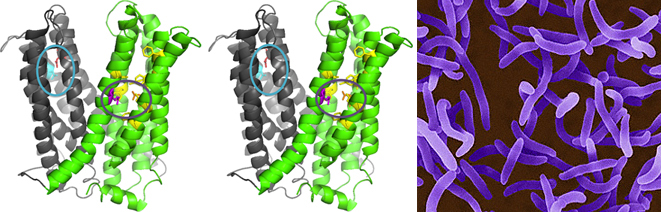
Figure 4. Structure model of outward-facing NorM from Vibrio cholerae. Stereoview (PDB 3MKT) showing TM1–6 in gray and TM7–12 in green. Asp36 (red) and neighboring Asn174 and Asn178 residues (light blue) are shown in a blue circle. Glu255 (purple) and Asp371 (orange) are shown in a purple circle with neighboring aromatic residues in yellow. Top, extracellular side; bottom, intracellular side. Panel on right-hand side: V. cholerae.