Investigation of activation-induced structural changes in ionotropic receptors using atomic force microscopy (AFM)
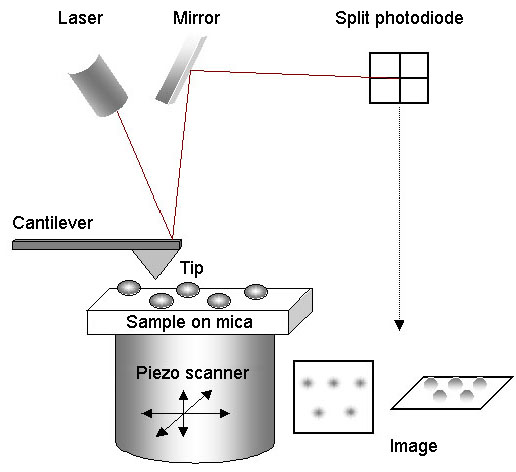
For AFM imaging, the sample is mounted on mica, and the tip is scanned across it in a raster pattern. A laser beam is bounced off the back of the cantilever and is reflected by a mirror onto a split photodiode. When the tip encounters an object on the surface of the mica, it becomes deflected, causing the position of the laser beam on the photodiode to change. The scanner moves in the Z-dimension to restore the beam to its original position. The XYZ movements of the scanner are analysed by a computer in order to generate the image. Importantly, imaging can be carried out under fluid, so that proteins can be studied under near-physiological conditions.
Several crystal and cryo-EM structures are available for ionotropic glutamate receptors (iGluRs). These studies provide fascinating 'snap-shots' of the receptors in different functional states. What is lacking, so far, is information about the kinetics underlying these structural transitions. We are using fast-scan atomic force microscopy (AFM) to study activation-induced structural changes in native, full-length iGluRs. We have found that activation triggers a rapid (<1-sec) and significant (~1-nm) reduction in the height of the extracellular domains of iGluRs integrated into supported lipid bilayers, and an increase in the relative mobility of the N-terminal domains. We are continuing to address the behaviour of iGluRs, and the effects of accessory proteins and drugs on these receptors. The figure shows the effect of activation on the height of an NMDA receptor.
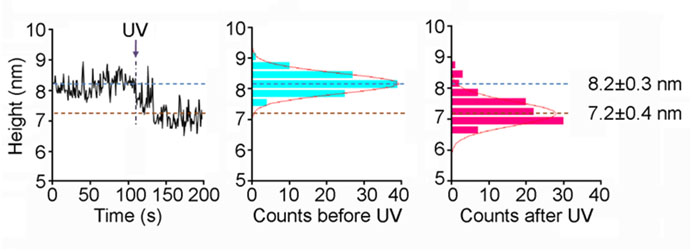
Fast-scan imaging of NMDA receptor height shifts. Frames were captured at 1 Hz. A single receptor was monitored in buffer containing 100 μM glycine and 100 μM caged-glutamate. UV irradiation occurred at 107 s. A fitted Gaussian curve is overlaid on each histogram.
Current lab member

PhD Student