Myc
Myc is a pleotropic transcription factor that serves as a pivotal, non-redundant instructor of tissue regeneration following injury. Myc expression in normal cells is tightly regulated. Following damage in a regenerative tissue, mitogenic stimulation leads to a transient increase in the short lived Myc protein and a burst of proliferation quickly follows. When Myc is deregulated it’s expression leads to uncontrolled cell cycle. Myc therefore plays a critical role in the progression and maintenance of tumours and is found deregulated in the vast majority of cancers. In addition to its cell intrinsic functions, Myc elicits tissue-specific transcriptional programmes that drive changes in the location and activity of stromal cells within the microenvironment.
The Wilson Lab uses acutely switchable models of Myc (MycERTM) to assess the immediate molecular and pathological effects of switching Myc on and off. Using these systems, we have shown that Myc binds to a large set of genes common to all tissues that are involved in cell cycle entry; suggesting Myc is able to drive cell cycle progression in any type of cell. However, despite acutely overexpressing Myc in all tissues, non-regenerative tissues like the heart remained almost entirely resistant to cell cycle entry, due to an inability of ectopic Myc to activate transcription of its target genes once bound. We have established that Myc driven transcription, and consequently cell proliferation, is critically dependent on the level of Cyclin T1 (Ccnt1) within a specific tissue.
The novel insights from our work opens up exciting new therapeutic approaches (i) to the inhibition of Myc-driven tumour growth by inhibiting Cyclin T1 and (ii) to the regeneration of damaged heart tissue by activating Myc transcription with Cyclin T1.
Inhibiting Myc
Cyclin T1 strongly associates with CDK9 to form the positive transcription elongation factor b (P-TEFb) that phosphorylates paused RNA Pol II, leading to transcriptional elongation.
Our data indicate Myc requires the maintained levels of Ccnt1 and Cdk9 to instruct its transcriptional program. Interestingly, Cyclin T1 protein level is a key factor in controlling the amount of the p-TEFb complex in a cell and overexpression of Ccnt1 in the heart leads to increased levels of both Cdk9 and phosphorylated RNA Pol II. As Myc frequently plays an obligate role as an oncogenic driver, targeting CCNT1 may be a selective and potent strategy to inhibit P-TEFb activity and suppress Myc-driven tumours. We aim to determine the effects of CCNT1 inhibition in cancers and develop novel CCNT1 inhibitors.
Regeneration
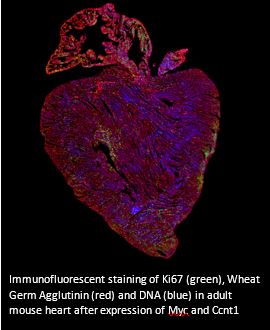 The adult heart is one of the least regenerative organs of the body. After mammalian birth, cardiomyocytes exit the cell cycle and subsequently turnover very slowly. Consequently, following injury the adult mammalian heart is unable to regenerate by replenishing lost cardiomyocytes, the default response of the adult mammalian heart is to replace the lost cardiomyocytes with non-contractile fibrotic scar tissue that ultimately results in heart failure. Using the heart as a model system, we overexpressed Ccnt1 specifically in the heart with an adeno associated virus. Together with our acutely switchable MycERTM system, we have demonstrated that overexpression of Ccnt1 is sufficient to enable Myc driven transcription. Once enabled, Myc transcriptional activation led to extensive cell cycle entry through to cytokinesis and a large increase in cardiomyocyte number and heart size.
The adult heart is one of the least regenerative organs of the body. After mammalian birth, cardiomyocytes exit the cell cycle and subsequently turnover very slowly. Consequently, following injury the adult mammalian heart is unable to regenerate by replenishing lost cardiomyocytes, the default response of the adult mammalian heart is to replace the lost cardiomyocytes with non-contractile fibrotic scar tissue that ultimately results in heart failure. Using the heart as a model system, we overexpressed Ccnt1 specifically in the heart with an adeno associated virus. Together with our acutely switchable MycERTM system, we have demonstrated that overexpression of Ccnt1 is sufficient to enable Myc driven transcription. Once enabled, Myc transcriptional activation led to extensive cell cycle entry through to cytokinesis and a large increase in cardiomyocyte number and heart size.
We aim to determine if cardiomyocyte proliferation after Myc activation and Ccnt1 expression can drive functional heart regeneration and understand the long-term effects to the heart.
Bywater, M. J. et al. & Wilson C.H. Reactivation of Myc transcription in the mouse heart unlocks its proliferative capacity. Nat. Comms. in press.