The main research interest of the Ladds group is to understand the molecular basis of agonist bias at G protein-coupled receptors. To accomplish this aim we use a range of multi-disciplinary approaches and model organisms.
We also collaborate extensively with global pharmaceutical companies in the pursuit of more efficacious and safer drugs (Academic working with AstraZeneca to develop safer drugs).
G protein-coupled receptors remain a leading drug target
G protein-coupled receptors (GPCRs) form the largest protein family in the human genome. ~30% of marketed drugs target these receptors and therefore understanding their signalling pathways is not simply an academic exercise
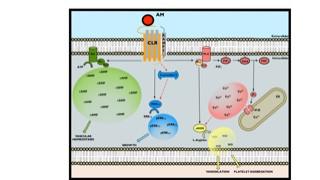
For many years it had been incorrectly assumed that GPCRs would elicit a single response to an agonist binding. However, there is now overwhelming evidence to suggest that many GPCRs exist in multiple receptor conformations and can elicit numerous functional responses, both G protein- and non G protein-dependent. Furthermore, agonists have the potential to activate the different signalling pathways to varying extents; a concept referred to as biased agonism (See figure for the adrenomedullin receptor).
The therapeutic promise of biased agonists is obvious; we could design ligands that actively engage with one beneficial signalling outcome while reducing the contribution observed at those that mediate more undesirable effects. Indeed, biased agonism probably explains why some drugs display clinical efficacy, while others fail, despite showing similar profiles of efficacy in preclinical trials.
GPCRs are numerous and share common structure
To date, over 800 different GPCRs have been suggested to exist within the human genome. The majority (~400) are involved with olfaction (specifically our abilities to smell different fragrances). The remaining non-olfactory GPCRs play a wide range of physiological roles including, but not limited to, glucose homeostasis, regulation of blood pressure and control of cognate function.
Unsurprisingly, many of these GPCRs have been targeted for therapeutic intervention. Examples include, anti-histamines, diabetic treatments, β-blockers, anti-psychotic drugs and many more. All GPCRs share a common architecture, consisting of 7 transmembrane domains, an extracellular N-terminus and an intracellular C-terminus.
The N-terminus is associated with binding of an extracellular ligand (frequently a hormone) while the intracellular loops (generated between the 7 transmembrane domains) transmit the signal to the associated heterotrimeric G protein. The activated GPCRs promotes dissociation of the heterotrimeric G protein enabling signal propagation.
GPCRs have been classified into different classes based upon their structure. The vast majority of mammalian GPCRs are found in Class A including the olfactory receptors.
Class B GPCRs are separated into 2 groups. Class B1 has 16 members and are characterised by binding peptide hormones and having a large (>120 amino acid) N-terminus. Class B2 are the so call adhesion receptors, who contain even larger N-terminal domains. Class C GPCRs are the so call venus-fly trap receptors, made up of large N-terminal domains and include receptors for taste. The Ladds group work on examples of GPCRs from all three classes.
Class A GPCRs

As part of our research, we focus upon developing and understanding the action of human adenosine receptors. Using a range of BRET-based assays we have identified novel adenosine receptor selective agonists (see BnOCPA as an example) and antagonists.
With our collaborators we are aiming to understand the mechanism of action of these novel compounds with a view to producing new adenosine receptor-based analgesics treatments for chronic pain relief. We are also using new methods to aid in the de-orphanisation (identification of endogenous agonists) for olfactory receptors.
Class B GPCRs

Some of the top drug targets for GPCRs can be found within the 16 members of the Class B1 subgroup. We are using BRET-based methods to characterise these peptide GPCRs. Specifically, we are interested in how a small group of molecular chaperonin proteins (RAMPs) control the pharmacology, trafficking and signalling output for these different receptors (See images of the glucagon and GLP-1 receptors).
Using cell-based assays ranging from model organism to primary human cells, we are characterising the features of the GPCRs, RAMPs and agonists that produce individual signalling outputs.
Class C GPCRs
This family of GPCRs have the distinction of being the first to demonstrate the concept of heterodimerisation. This was shown for the GABAB receptor. We now know that other members of this group also share this ability to dimerise.
We are focused upon the different complexes formed for the taste receptors. These receptors enable us to sense sweetness or bitterness etc. We are exploring the different signalling mechanisms used by taste receptors with an overall goal of producing tuned glucose sensing receptors.
Using mathematics to complement our studies
While pharmacological experimentation can provide a wealth of kinetic data, ranging from ligand-binding, to receptor internalisation, it is almost impossible to acquire dynamic rates for each individual reaction and species.
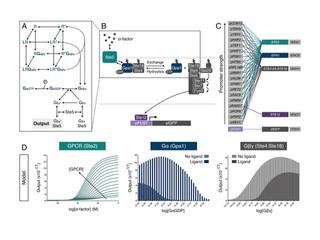
It has become apparent that mathematical modelling can be used to provide a more complete understanding of cell signalling dynamics in a pharmacological setting by ‘filling in’ the gaps in experimental work (see figure from Shaw et al.).
We are using mathematical models to help understand how drugs targeting GPCRs can activate multiple signalling pathways potential leading to unwanted side effects.